PLoS has made it’s first foray into ‘book’ publishing, with it’s new collection from PLOS Computational Biology called Translational Bioinformatics.
I think this is a great idea, and as founding editor Phil Bourne points out, are a far better option for both readers and authors than the ‘traditional’ science books made up of contributed chapters, which are hard to access, expensive and rarely cited.
Of particular interest to us in the microbial world is Chapter 12: Human Microbiome Analysis by Xochitl C. Morgan, Curtis Huttenhower from Harvard.
Being a book-chapter-style document, it includes some really useful ‘extras’ including questions to get you thinking about what you’ve learnt from the article (and answers in the supplementary), a useful glossary and pointers for further reading.
Because it is PLoS, I can show you exactly what is in it:
Abstract
Humans are essentially sterile during gestation, but during and after birth, every body surface, including the skin, mouth, and gut, becomes host to an enormous variety of microbes, bacterial, archaeal, fungal, and viral. Under normal circumstances, these microbes help us to digest our food and to maintain our immune systems, but dysfunction of the human microbiota has been linked to conditions ranging from inflammatory bowel disease to antibiotic-resistant infections. Modern high-throughput sequencing and bioinformatic tools provide a powerful means of understanding the contribution of the human microbiome to health and its potential as a target for therapeutic interventions. This chapter will first discuss the historical origins of microbiome studies and methods for determining the ecological diversity of a microbial community. Next, it will introduce shotgun sequencing technologies such as metagenomics and metatranscriptomics, the computational challenges and methods associated with these data, and how they enable microbiome analysis. Finally, it will conclude with examples of the functional genomics of the human microbiome and its influences upon health and disease.
Citation: Morgan XC, Huttenhower C (2012) Chapter 12: Human Microbiome Analysis. PLoS Comput Biol 8(12): e1002808. doi:10.1371/journal.pcbi.1002808
What to Learn in This Chapter
- An overview of the analysis of microbial communities
- Understanding the human microbiome from phylogenetic and functional perspectives
- Methods and tools for calculating taxonomic and phylogenetic diversity
- Metagenomic assembly and pathway analysis
- The impact of the microbiome on its host
1. Introduction
2. A Brief History of Microbiome Studies
3. Taxonomic Diversity
4. Shotgun Sequencing and Metagenomics
5. Computational Functional Metagenomics
6. Host Interactions and Interventions
7. Summary
8. Exercises
Supporting Information
Acknowledgments
References
Figures
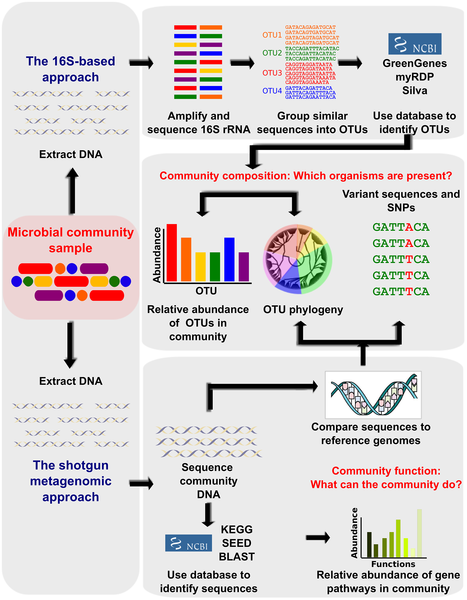
Figure 1. Bioinformatic methods for functional metagenomics.
Studies that aim to define the composition and function of uncultured microbial communities are often referred to collectively as “metagenomic,” although this refers more specifically to particular sequencing-based assays. First, community DNA is extracted from a sample, typically uncultured, containing multiple microbial members. The bacterial taxa present in the community are most frequently defined by amplifying the 16S rRNA gene and sequencing it. Highly similar sequences are grouped into Operational Taxonomic Units (OTUs), which can be compared to 16S databases such as Silva, Green Genes, and RDP to identify them as precisely as possible. The community can be described in terms of which OTUs are present, their relative abundance, and/or their phylogenetic relationships. An alternate method of identifying community taxa is to directly metagenomically sequence community DNA and compare it to reference genomes or gene catalogs. This is more expensive but provides improved taxonomic resolution and allows observation of single nucleotide polymorphisms (SNPs) and other variant sequences. The functional capabilities of the community can also be determined by comparing the sequences to functional databases (e.g. KEGG or SEED). This allows the community to be described as relative abundances of its genes and pathways.
doi:10.1371/journal.pcbi.1002808.g001

Figure 2. Ecological representations of microbial communities: collector’s curves, alpha, and beta diversity.
These examples describe the A) sequence counts and B) relative abundances of six taxa (A, B, C, D, E, and F) detected in three samples. C) A collector’s curve, typically generated using a richness estimator such as Chao1 or ACE, approximates the relationship between the number of sequences drawn from each sample and the number of taxa expected to be present based on detected abundances. D) Alpha diversity captures both the organismal richness of a sample and the evenness of the organisms’ abundance distribution. Here, alpha diversity is defined by the Shannon index, where pi is the relative abundance of taxon i, although many other alpha diversity indices may be employed. E) Beta diversity represents the similarity (or difference) in organismal composition between samples. In this example, it can be simplistically defined by the equation , where n1 and n2 are the number of taxa in samples 1 and 2, respectively, and c is the number of shared taxa, but again many metrics such as Bray-Curtis or UniFrac are commonly employed.
doi:10.1371/journal.pcbi.1002808.g002
Happy reading!
